
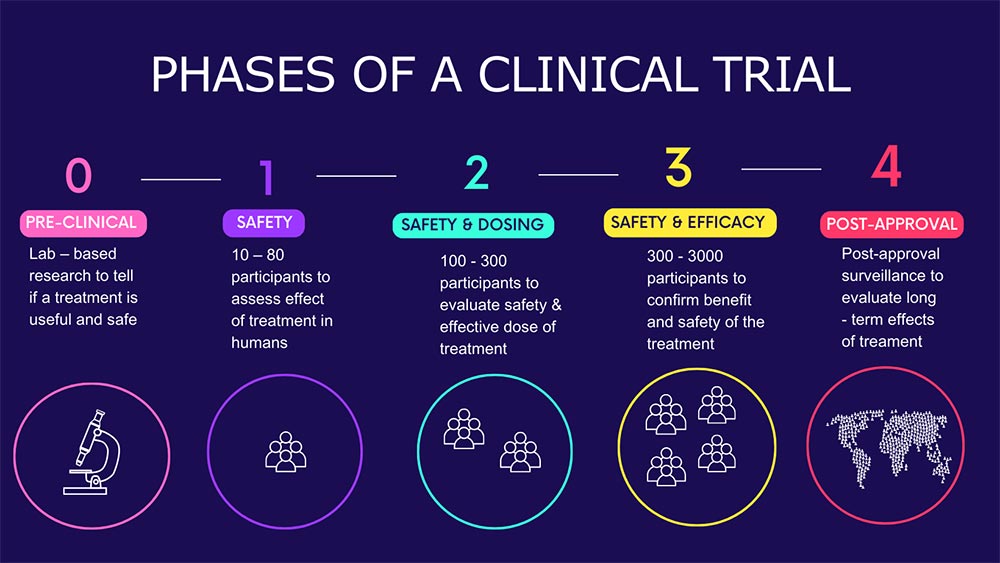
Bringing a new therapy to market involves one of the most rigorously structured processes in modern medicine. According to a study published in PubMed Central, nine out of ten drug candidates that enter clinical studies fail during Phase I, II, or III trials, or at the regulatory approval stage. That statistic alone reflects the scientific, operational, and regulatory complexity embedded across every phase of development.
For clinical development teams managing multi-site or multi-country studies, fragmented execution across planning, monitoring, safety reporting, and data management is one of the most persistent sources of delay and protocol deviation. Understanding the End-to-End Clinical Trial Process from protocol development through Clinical Study Report (CSR) submission gives sponsors and operations leaders the framework to anticipate risks, allocate resources accurately, and maintain regulatory alignment throughout.
This overview covers each stage of the clinical trial lifecycle with the operational and regulatory depth required to manage trials effectively, particularly for Phase II and Phase III programs targeting United States Food and Drug Administration (FDA) and European Medicines Agency (EMA) acceptance.
Why an End-to-End View of the Clinical Trial Process Matters?
Most trial failures are not caused by a single catastrophic event. They accumulate from coordination gaps between phases, insufficient planning for regulatory timelines, and reactive rather than proactive safety and data management. A stage-by-stage view of the full trial process addresses this by establishing clear dependencies between each phase.
According to the Biotechnology Innovation Organization (BIO), the average likelihood of approval for all drugs in Phase I development is approximately 7.9%, and the average time to regulatory approval from Phase I is 10.5 years. These figures indicate that success is rarely a function of scientific potential alone, and operational discipline across the entire trial lifecycle determines whether a program advances or stalls.
A BIO study analyzing 9,704 clinical development programs over 2011–2020 found Phase II-to-III success at 28.9% and Phase III-to-approval at 57.8%, underscoring the importance of structured, compliance-driven execution at each stage.
The sections below map each stage of the trial process with the operational and regulatory requirements that determine whether a program moves forward or faces delay.
The End-to-End Clinical Trial Process
A clinical trial is not a linear sequence of independent activities. It is a set of interdependent functions, each of which must satisfy both scientific and regulatory standards before the next stage can proceed. The following breakdown covers the key stages from protocol development through submission readiness.
1. Clinical Trial Planning and Protocol Development
Every trial begins with a study protocol defining objectives, eligibility criteria, treatment regimens, endpoints, safety reporting, and the Statistical Analysis Plan (SAP).
Protocol development requires early alignment across clinical operations, regulatory, biostatistics, and pharmacovigilance teams. Poor protocol design is a leading cause of deviations, non-compliance, and data integrity issues during trial conduct.
At this stage, sponsors finalize the study design and regulatory pathway (e.g., IND or CTA), ensuring full compliance with ICH-GCP requirements from the outset.
2. Site Feasibility and Selection
Site feasibility directly impacts recruitment speed, data quality, and protocol adherence. Assessments typically evaluate:
- Investigator experience and site performance history.
- Patient availability and recruitment potential.
- Infrastructure, staffing, and EDC readiness.
- Local regulatory and ethics timelines.
- Standard-of-care and cultural considerations.
For multi-country studies, feasibility must account for country-specific import regulations, language requirements, and variability in ethics review. Inadequate feasibility planning is a common cause of downstream enrollment delays.
3. Regulatory and Ethics Approvals
Before patient enrollment begins, all applicable regulatory and ethics approvals must be secured. The approvals required vary by country and trial type, but typically include the following:
| Approval Type | Applicable Body | Scope |
| IND Application | US FDA | Authorization to initiate human trials in the US. |
| CTA Submission | EMA / National Competent Authorities. | Trial authorization across EU member states. |
| Institutional Review Board / Ethics Committee (IRB/EC) Approval | Local / Central IRB or EC | Ethical conduct and participant protection. |
| Drug Import License / No Objection Certificate (NOC) | Country-Specific Regulatory Authority. | Required for IMP shipment into the study country. |
| Trial Master File (TMF) Setup | Site and Sponsor. | Repository for all essential trial documents. |
In multi-regional trials, regulatory timelines vary significantly by jurisdiction. Delays in ethics approval or IMP import clearance in one country can affect the entire enrollment timeline if contingency planning was not built into the project schedule. Risk-based project management, which maps alternative approval pathways and timelines in advance, reduces the likelihood of cascading delays.
4. Study Start-Up and Site Initiation
Study start-up (SSU) covers the transition from approval to first patient enrolled and is often a major timeline bottleneck. Key activities include:
- Site initiation visits and protocol training.
- Confirmation of regulatory readiness.
- IMP delivery and verification.
- TMF activation and documentation finalization.
- Pharmacovigilance system setup.
High-quality SSU is critical. Poorly prepared sites consistently show higher protocol deviation and data discrepancy rates.
5. Patient Recruitment and Enrollment
Patient recruitment is the single most common cause of clinical trial delays. Enrollment shortfalls extend study timelines, increase costs, and, in some cases, require protocol amendments to adjust eligibility criteria, thereby introducing additional regulatory review cycles.
Effective recruitment requires:
- Feasibility-driven patient identification strategies.
- Site-level enrollment targets and tracking.
- Referral networks and patient support programs.
- Real-time CTMS oversight.
- Decentralized and remote trial capabilities.
6. Trial Conduct and Monitoring
Once sites are initiated and patients enrolled, ongoing monitoring ensures that trial conduct remains compliant with the protocol, ICH-GCP guidelines, and sponsor-defined quality standards.
Modern clinical trials typically combine on-site monitoring with central and remote monitoring in a hybrid model:
- On-site monitoring involves Clinical Research Associates (CRAs) visiting sites to verify source data against case report forms (CRFs), confirm protocol adherence, and assess site performance.
- Central monitoring uses real-time data analysis, statistical signals, and risk-based triggers to identify protocol deviations, data anomalies, and site-level risks without requiring an immediate site visit.
- Remote monitoring tools, including secure electronic access to site data, allow for continuous oversight between on-site visits.
7. Safety Monitoring and Pharmacovigilance
Safety oversight runs continuously from the first patient enrolled through the final reporting. Core activities include:
- SAE and SUSAR detection and reporting.
- Signal detection and risk evaluation.
- DSMB or IDMC reporting where required.
- Maintenance of audit-ready safety databases.
SUSAR timelines are strictly regulated, and non-compliance can trigger clinical holds or broader regulatory scrutiny.
8. Clinical Data Management and Review
Clinical Data Management (CDM) ensures data integrity from collection through database lock. Key activities include:
- EDC build and validation.
- Data entry, query management, and SDV.
- Medical coding (e.g., MedDRA, WHO Drug).
- Ongoing data quality review and audit trail maintenance.
Non-compliant data systems or audit gaps are a common cause of regulatory review delays.
9. Study Close-Out and Database Lock
After Last Patient Last Visit (LPLV), close-out activities focus on operational and data finalization, including:
- Site close-out visits and IMP reconciliation.
- Final query resolution.
- Safety data reconciliation.
- TMF review and archiving.
- Database lock approval.
Once locked, data changes require formal justification and re-lock procedures.
10. Clinical Study Reporting and Submission Readiness
The Clinical Study Report (CSR) summarizes trial design, conduct, efficacy, and safety in compliance with ICH E3 guidelines. Submission readiness includes:
- Final statistical analysis per SAP.
- CSR authoring and review.
- eCTD-compliant filing preparation.
Any unresolved data, documentation gaps, or TMF deficiencies at this stage can delay submission. Effective submission planning must begin well before LPLV.
Key Factors That Influence Successful End-to-End Trial Execution
The clinical trial process does not fail in isolation at a single stage. Failure patterns compound across phases when the following operational and regulatory factors are not adequately addressed from the outset:
| Factor | Impact on Trial Outcomes |
| Protocol quality and operational feasibility | Directly affects site compliance, deviation rates, and regulatory acceptance. |
| Site feasibility rigor | Determines recruitment velocity and data quality before enrollment begins. |
| Regulatory and ethics planning by country | Prevents SSU delays and IMP logistics failures. |
| Risk-based monitoring framework | Reduces protocol deviations and accelerates database lock. |
| Pharmacovigilance system integration | Ensures SUSAR reporting compliance and safety data submission-readiness. |
| Data management infrastructure | Determines whether the final dataset will withstand FDA/EMA scrutiny. |
| Vendor and service coordination | Fragmented vendor management is a primary driver of cost overruns and timeline slippage. |
For Phase II and Phase III trials operating across multiple countries, an additional factor is maintaining consistent operational standards across geographically diverse sites with different regulatory environments, standard-of-care practices, and patient enrollment characteristics. End-to-end accountability, from a single operational command center that manages all functions under a single governance framework, reduces the coordination risk inherent in fragmented, multi-vendor models.
Conclusion
The end-to-end clinical trial process spans protocol development, regulatory approvals, site operations, patient enrollment, safety management, data management, and submission readiness. Each stage is sequentially and operationally dependent on the stages that precede it.
Given that over 90% of drug candidates entering clinical trials do not reach regulatory approval, the margin for operational error is narrow. Trials that fail to meet recruitment timelines, generate clean, audit-ready data, or maintain ICH-GCP-compliant pharmacovigilance documentation face not only timeline and cost consequences but also the risk of regulatory rejection of otherwise scientifically valid programs.
For sponsors managing Phase II and Phase III programs, structuring trial operations with full-service, end-to-end oversight, from feasibility through CSR submission, remains the most effective strategy for protecting both timeline and regulatory investment.
